Internals¶
Overview¶
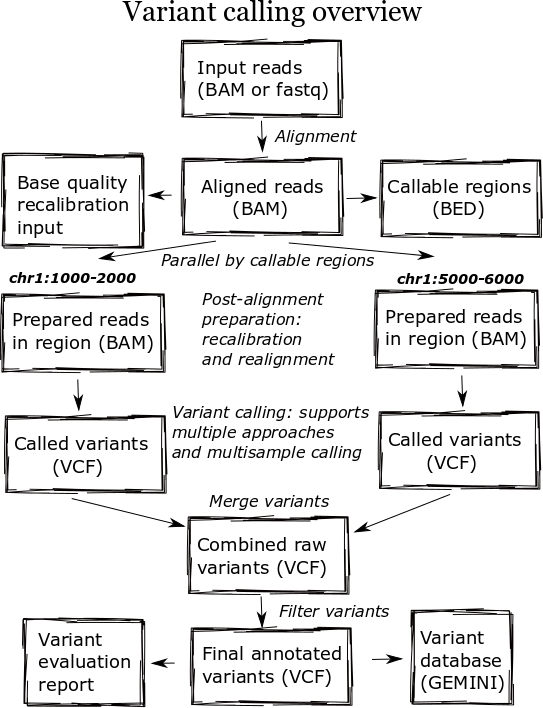
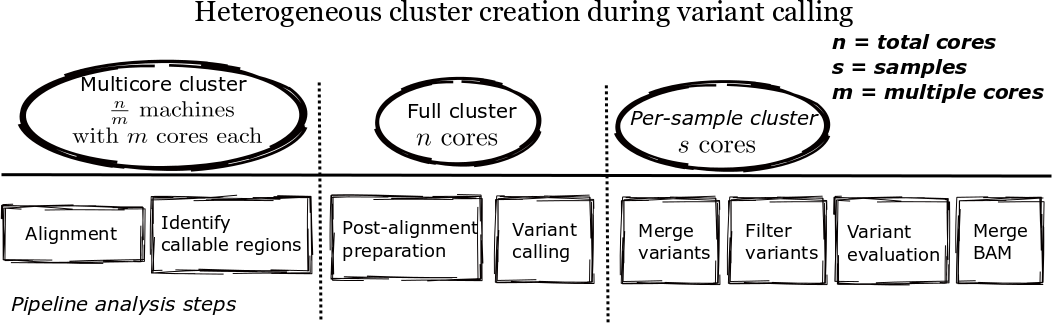
Parallel¶
bcbio calculates callable regions following alignment using goleft depth. These regions determine breakpoints for analysis, allowing parallelization by genomic regions during variant calling. Defining non-callable regions allows bcbio to select breakpoints for parallelization within chromosomes where we won’t accidentally impact small variant detection. The callable regions also supplement the variant calls to define positions where not called bases are homozygous reference, as opposed to true no-calls with no evidence. The callable regions plus variant calls is an alternative to gVCF output which explicitly enumerates reference calls in the output variant file.
Somatic tumor only variant calling pipeline with UMIs step by step¶
Minor steps (like tabix’ing of vcfs, indexing of bams) and details (full paths) are omitted.
bcbio.yaml config:
details:
- algorithm:
aligner: bwa
trim_ends: [2,0,2,0]
min_allele_fraction: 0.01
correct_umis: /path/umi.whitelist.txt
tools_off:
- gemini
umi_type: fastq_name
variantcaller:
- vardict
coverage: target.bed
variant_regions: target.bed
analysis: variant2
description: samplex
files:
- /path/samplex_1.fq.gz
- /path/samplex_2.fq.gz
genome_build: hg38
metadata:
phenotype: tumor
resources:
fgbio:
options: [--min-reads, 3]
trimming 2p from reads: trim_ends: [2,0,2,0], indexing:
bgzip --threads 8 -c <(seqtk trimfq -b 2 -e 0 samplex_1.fq.gz) > samplex_1.fq.gz bgzip --threads 8 -c <(seqtk trimfq -b 2 -e 0 samplex_2.fq.gz) > samplex_2.fq.gz grabix index samplex_2.fq.gz grabix index samplex_1.fq.gz
alignment with bwa mem, sort and mark duplicates, assign BAM tags (XS=sample, XC=cell, RX=UMI), input: fastq files, output: samplex-sort.bam:
unset JAVA_HOME && \ bwa mem -c 250 -M -t 16 -R '@RG\tID:samplex\tPL:illumina\tPU:samplex\tSM:samplex' \ -v 1 /path/reference/genomes/Hsapiens/hg38/bwa/hg38.fa samplex_1.fq.gz samplex_2.fq.gz | \ bamsormadup tmpfile=samplex-sort-sorttmp-markdup inputformat=sam threads=16 \ outputformat=bam level=0 SO=coordinate | \ /path/bcbio/anaconda/envs/python2/bin/python /path/bin/umis bamtag - | \ samtools view -b > samplex-sort.bam
correct UMIs with fgbio using the whitelist, input: samplex-sort.bam, output: samplex-sort-umis_corrected.bam:
unset JAVA_HOME && \ fgbio -Xms750m -Xmx30g -XX:+UseSerialGC --tmp-dir . --async-io=true --compression=0 \ CorrectUmis \ -t XC -m 3 -d 1 -x -U /path/umi.whitelist.txt -i samplex-sort.bam \ -o samplex-sort-umis_corrected.bam
calculate coverage with mosdepth:
mosdepth -t 16 -F 1804 --no-per-base --by target.bed samplex-rawumi \ samplex-sort-umis_corrected.bam
fgbio GroupReadsByUmi, CallDuplexConsensusReads, FilterConsensusReads with min-reads=3, bam2fastq:
unset JAVA_HOME && \ fgbio -Xms750m -Xmx30g -XX:+UseSerialGC --tmp-dir . --async-io=true --compression=0 \ GroupReadsByUmi \ --edits=1 --min-map-q=1 -t XC -s paired -i samplex-sort-umis_corrected.bam | \ fgbio -Xms750m -Xmx30g -XX:+UseSerialGC --tmp-dir . --async-io=true --compression=0 \ CallDuplexConsensusReads \ --min-input-base-quality=2 --sort-order=:none: -i /dev/stdin -o /dev/stdout | \ fgbio -Xms750m -Xmx30g -XX:+UseSerialGC --tmp-dir . --async-io=true --compression=0 \ FilterConsensusReads --min-reads=3 --min-base-quality=13 --max-base-error-rate=0.1 \ -r /path/reference/genomes/Hsapiens/hg38/seq/hg38.fa -i /dev/stdin -o /dev/stdout | \ bamtofastq collate=1 T=samplex-sort-umis_corrected-cumi-1-bamtofastq-tmp \ F=samplex-sort-umis_corrected-cumi-1.fq.gz F2=samplex-sort-umis_corrected-cumi-2.fq.gz tags=cD,cM,cE gz=1
align consensus reads:
unset JAVA_HOME && bwa mem -C -c 250 -M -t 16 -R '@RG\tID:samplex\tPL:illumina\tPU:samplex\tSM:samplex' \ -v 1 /projects/ngs/reference/genomes/Hsapiens/hg38/bwa/hg38.fa \ samplex-sort-umis_corrected-cumi-1.fq.gz samplex-sort-umis_corrected-cumi-2.fq.gz | \ samtools sort -@ 16 -m 1G -T samplex-sort-cumi-sorttmp -o samplex-sort-cumi.bam /dev/stdin samtools index -@ 16 samplex-sort-cumi.bam samplex-sort-cumi.bam.bai
clean variant_regions bed file:
cat target.bed | grep -v ^track | grep -v ^browser | grep -v ^@ | grep -v ^# | \ bcbio_python -c 'from bcbio.variation import bedutils; bedutils.remove_bad()' | \ sort -V -T . -k1,1 -k2,2n > cleaned-target.bed cat cleaned-target.bed | bgzip --threads 16 -c > cleaned-target.bed.gz tabix -f -p bed cleaned-target.bed.gz bedtools merge -i cleaned-target.bed> cleaned-target-merged.bed cat cleaned-target-merged.bed | bgzip --threads 16 -c > cleaned-target-merged.bed.gz
clean coverage bed file (the same in our example):
cat target.bed | grep -v ^track | grep -v ^browser | grep -v ^@ | grep -v ^# | \ iconv -c -f utf-8 -t ascii | sed 's/ //g' | \ bcbio_python -c 'from bcbio.variation import bedutils; bedutils.remove_bad()' | \ sort -V -T . -k1,1 -k2,2n > cov-target.bed cat cov-target.bed | bgzip --threads 16 -c > cov-target.bed.gz tabix -f -p bed cov-target.bed.gz bedtools merge -i cov-target.bed > cov-target-merged.bed cat cov-target-merged.bed | bgzip --threads 16 -c > cov-target-merged.bed.gz
clean sv regions bed file:
cat cleaned-target.bed | grep -v ^track | grep -v ^browser | grep -v ^@ | grep -v ^# | \ /home/kmhr378/local/bin/bcbio_python -c 'from bcbio.variation import bedutils; bedutils.remove_bad()' | \ sort -V -T . -k1,1 -k2,2n > \ svregions-cleaned-target.bed cat svregions-cleaned-target.bed | bgzip --threads 16 -c > svregions-cleaned-target.bed.gz
calculate coverage for 3 bed files with MOSDEPTH:
export MOSDEPTH_Q0=NO_COVERAGE && export MOSDEPTH_Q1=LOW_COVERAGE && \ export MOSDEPTH_Q2=CALLABLE && \ mosdepth -t 16 -F 1804 -Q 1 --no-per-base --by cleaned-target.bed \ --quantize 0:1:4: samplex-variant_regions samplex-sort-cumi.bam mosdepth -t 16 -F 1804 --no-per-base --by svregions-cleaned-target.bed \ samplex-sv_regions samplex-sort-cumi.bam mosdepth -t 16 -F 1804 --no-per-base --by cov-target.bed samplex-coverage \ samplex-sort-cumi.bam \ -T 1,5,10,20,50,100,200,500,1000,2000,5000,10000,20000,50000,100000,200000,500000
hts_nim counts:
hts_nim_tools count-reads -t 16 -F 1804 /path/samplex/counts/fullgenome.bed samplex-sort-cumi.bam > fullgenome-1804-counts.txt hts_nim_tools count-reads -t 16 -F 1804 cleaned-target.bed samplex-sort-cumi.bam > cleaned-target-merged-1804-counts.txt
samtools read statistics::
samtools stats -@ 16 samplex-sort-cumi.bam > samplex.txt samtools idxstats samplex-sort-cumi.bam > samplex-idxstats.txt
variant calling with vardict (repeated for each alignment chunk):
unset R_HOME && unset R_LIBS && unset JAVA_HOME && \ export VAR_DICT_OPTS='-Xms750m -Xmx3500m -XX:+UseSerialGC -Djava.io.tmpdir=.' && \ vardict-java -G /path/reference/genomes/Hsapiens/hg38/seq/hg38.fa \ -N samplex -b samplex-sort-cumi.bam -c 1 -S 2 -E 3 -g 4 --nosv --deldupvar -Q 10 -F 0x700 \ samplex-chr5_0_x-unmerged-regions-regionlimit.bed -f 0.0025 | \ teststrandbias.R | \ var2vcf_valid.pl -A -N samplex -E -f 0.0025 | grep -v ^##contig | \ bcftools annotate -h samplex-chr5_0_x-contig_header.txt | \ bcftools filter -i 'QUAL >= 0' | \ bcftools filter --soft-filter 'LowFreqBias' --mode '+' -e 'FORMAT/AF[0:0] < 0.02 && \ FORMAT/VD[0] < 30 && INFO/SBF < 0.1 && INFO/NM >= 2.0' | \ awk -F$'\t' -v OFS='\t' '{if ($0 !~ /^#/) gsub(/[KMRYSWBVHDXkmryswbvhdx]/, "N", $4) } {print}' | \ awk -F$'\t' -v OFS='\t' '{if ($0 !~ /^#/) gsub(/[KMRYSWBVHDXkmryswbvhdx]/, "N", $5) } {print}' | \ awk -F$'\t' -v OFS='\t' '$1!~/^#/ && $4 == $5 {next} {print}' | \ vcfstreamsort | bgzip -c > samplex-chr5_0_x.vcf.gz zgrep ^# samplex-chr5_0_x.vcf.gz > samplex-chr5_0_x-fixheader-header.vcf unset JAVA_HOME && \ picard FixVcfHeader HEADER=samplex-chr5_0_x-fixheader-header.vcf \ INPUT=samplex-chr5_0_x.vcf.gz \ OUTPUT=samplex-chr5_0_x-fixheader.vcf.gz
gather vcfs:
unset JAVA_HOME && \ gatk --java-options -Xms681m -Xmx3181m -XX:+UseSerialGC -Djava.io.tmpdir=. \ GatherVcfs -I samplex-files.list -O samplex.vcf.gz
annotate with snpEff:
unset JAVA_HOME && \ snpEff -Xms750m -Xmx29g -Djava.io.tmpdir=. eff \ -dataDir /path/reference/genomes/Hsapiens/hg38/snpeff \ -hgvs -cancer -noLog -i vcf -o vcf -csvStats samplex-effects-stats.csv \ -s samplex-effects-stats.html GRCh38.86 samplex.vcf.gz | \ bgzip --threads 16 -c > samplex-effects.vcf.gz
annotate with vcfanno:
vcfanno -p 16 dbsnp.conf samplex-effects.vcf.gz | \ bcftools reheader -h samplex-effects-annotated-sample_header.txt | \ bcftools view | bgzip -c > samplex-effects-annotated.vcf.gz tabix -f -p vcf samplex-effects-annotated.vcf.gz vcfanno -p 16 samplex-effects-annotated-annotated-somatic-combine.conf \ samplex-effects-annotated.vcf.gz | bgzip -c > samplex-effects-annotated-annotated-somatic.vcf.gz tabix -f -p vcf samplex-effects-annotated-annotated-somatic.vcf.gz cat samplex-effects-annotated-annotated-somatic-priority.tsv | bgzip --threads 16 -c > \ samplex-effects-annotated-annotated-somatic-priority.tsv.gz tabix -f -0 -c '#' -s 1 -b 2 -e 3 samplex-effects-annotated-annotated-somatic-priority.tsv.gz
Tests¶
To run bcbio automated tests, install bcbio and clone bcbio master repository. You are testing your installation with tests provided in bcbio-nextgen/tests:
which bcbio_nextgen.py
cd bcbio-nextgen/tests
./run_tests.sh > tests.out
Tests are in integration/*.py. Each test has a set or marks. Marks are listed in pytest.ini. The mark defines how many tests to select. By default (just running plain ./run_tests.sh), it is speed1 = 11 tests.